La Educación en la Química en Argentina y en el Mundo
EL PREMIO NOBEL DE QUÍMICA 2024
Traducción de Luz Lastres
Universidad de Buenos Aires
E-mail: klastres@gmail.com
Recibido: 06/12/2024. Aceptado: 15/12/2024.
Resumen. Se presenta una traducción al español de artículos difundidos en la página web del Premio Nobel. En esta traducción se incluye información referente a las investigaciones realizadas en el diseño computacional y la predicción de la estructura de las proteínas de David Baker, Demis Hassabis y John Jumper, por las cuales fueron distinguidos por el Premio Nobel en Química en 2024. Además, se realizó una edición y actualización del texto por parte de la directora de la revista.
Palabras clave. Premio Nobel, diseño computacional, predicción de estructuras, proteínas, inteligencia artificial.
The Nobel Prize in Chemistry 2024
Abstract. A Spanish translation of articles disseminated on the Nobel Prize website is presented. This translation includes information regarding the research conducted in computational design and the prediction of protein structures by David Baker, Demis Hassabis, and John Jumper, for which they were awarded the Nobel Prize in Chemistry in 2024. Additionally, the text was edited and updated by the magazine's editor-in-chief.
Keywords. Nobel prize, computational design, structure prediction, proteins, artificial intelligence.
Traducción al español de información publicada por la Real Academia Sueca de Ciencias:
Science Editors: Peter Brzezinski, Heiner Linke, Johan Åqvist, the Nobel Committee for Chemistry
Text: Ann Fernholm
Translation: Clare Barnes
Illustrations: Johan Jarnestad, Terezia Kovalova.
Editor: Vincent von Sydow
©The Royal Swedish Academy of Sciences
Edición de la traducción al español: María Gabriela Lorenzo
La Real Academia Sueca de Ciencias ha decidido otorgar el Premio Nobel de Química 2024, con una mitad a DAVID BAKER, “por el diseño computacional de proteínas”; y, la otra mitad de manera conjunta a DEMIS HASSABIS y JOHN JUMPER, “por la predicción de la estructura de las proteínas”.
![]()
 Estos investigadores han
revelado los secretos de las proteínas a través de la computación y la
inteligencia artificial.
Estos investigadores han
revelado los secretos de las proteínas a través de la computación y la
inteligencia artificial.
Los químicos llevan mucho tiempo soñando con comprender y dominar por completo las herramientas químicas de la vida: las proteínas. Este sueño ahora está a nuestro alcance. Demis Hassabis y John Jumper han utilizado con éxito la inteligencia artificial para predecir la estructura de casi todas las proteínas conocidas. David Baker ha aprendido a dominar los componentes básicos de la vida y a crear proteínas completamente nuevas. El potencial de sus descubrimientos es enorme.
¿Cómo es posible la exuberante química de la vida? La respuesta a esta pregunta es la existencia de las proteínas, que pueden describirse como brillantes herramientas químicas. Generalmente están formadas por veinte aminoácidos que se pueden combinar de infinitas maneras. Utilizando la información almacenada en el ADN como modelo, los aminoácidos se unen entre sí en nuestras células para formar largas cadenas.
Entonces ocurre la magia de las proteínas: la cadena de aminoácidos se retuerce y se pliega en una estructura tridimensional distintiva, a veces única (Figura 1). Esta estructura es la que da a las proteínas su función. Algunas se convierten en bloques químicos que pueden crear músculos, cuernos o plumas, mientras que otras pueden convertirse en hormonas o anticuerpos. Muchas de ellas forman enzimas, que impulsan las reacciones químicas de la vida con una precisión asombrosa. Las proteínas que se encuentran en las superficies de las células también son importantes y funcionan como canales de comunicación entre la célula y su entorno.
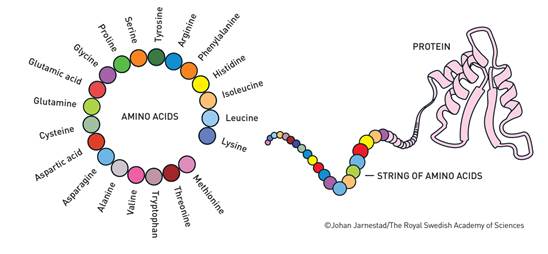
Figura 1. Aminoácidos y estructura de las proteínas
Es difícil exagerar el potencial que encierran los componentes químicos de la vida, estos veinte aminoácidos. El Premio Nobel de Química 2024 es sobre comprenderlos y dominarlos a un nivel completamente nuevo. La mitad del premio se otorga a Demis Hassabis y John Jumper, quienes han utilizado inteligencia artificial para resolver con éxito un problema con el que los químicos han luchado durante más de cincuenta años: predecir la estructura tridimensional de una proteína a partir de una secuencia de aminoácidos. Esto les ha permitido predecir la estructura de casi todos los 200 millones de proteínas conocidas. La otra mitad del premio se otorga a David Baker. Ha desarrollado métodos informáticos para lograr lo que mucha gente creía que era imposible: crear proteínas que no existían anteriormente y que, en muchos casos, tienen funciones completamente nuevas.
El Premio Nobel de Química 2024 reconoce dos descubrimientos diferentes, pero, como se verá, están estrechamente relacionados. Para entender los desafíos que han superado los galardonados de este año, debemos remontarnos a los albores de la bioquímica moderna
Las primeras imágenes granuladas de proteínas
Los químicos saben desde el siglo XIX que las proteínas son importantes para los procesos vitales, pero hubo que esperar hasta la década de 1950 para que las herramientas químicas fueran lo suficientemente precisas como para que los investigadores comenzaran a explorar las proteínas con más detalle. Los investigadores de Cambridge, John Kendrew y Max Perutz (Figura 2) hicieron un descubrimiento revolucionario cuando, a finales de la década, utilizaron con éxito la cristalografía de rayos X para presentar los primeros modelos tridimensionales de proteínas. En reconocimiento a este descubrimiento, recibieron el Premio Nobel de Química en 1962. Posteriormente, los investigadores han utilizado principalmente la cristalografía de rayos X, y a menudo un gran esfuerzo, para producir con éxito imágenes de alrededor de 200.000 proteínas diferentes, lo que sentó las bases para el Premio Nobel de Química de 2024.


Figura 2. John Kendrew y Max Perutz
Un acertijo: ¿Cómo encuentra una proteína su estructura única?
Christian Anfnsen (Figura 3), un científico estadounidense, hizo otro descubrimiento temprano. Utilizando varios trucos químicos, logró hacer que una proteína existente se desplegara y luego se plegara de nuevo. La observación interesante fue que la proteína adoptó exactamente la misma forma cada vez. En 1961, concluyó que la estructura tridimensional de una proteína está completamente gobernada por la secuencia de aminoácidos en la proteína. Esto lo llevó a ser galardonado con el Premio Nobel de Química en 1972.
Sin embargo, la lógica de Anfnsen contiene una paradoja, que otro estadounidense, Cyrus Levinthal (Figura 3), señaló en 1969. Calculó que incluso si una proteína solo consta de 100 aminoácidos, en teoría la proteína puede asumir al menos 1047 estructuras tridimensionales diferentes. Si la cadena de aminoácidos se plegara al azar, se necesitaría más tiempo que la edad del universo para encontrar la estructura proteica correcta. En una célula, solo se necesitan unos pocos milisegundos. Entonces, ¿cómo se pliega realmente la cadena de aminoácidos?
El descubrimiento de Anfnsen y la paradoja de Levinthal implicaron que el plegamiento es un proceso predeterminado. Y, lo que es más importante, toda la información sobre cómo se pliega la proteína debe estar presente en la secuencia de aminoácidos.


Figura 3. Christian Anfnsen y Cyrus Levinthal
Arrojando el guante al gran desafío de la bioquímica
Los resultados anteriores condujeron a otra conclusión decisiva: si los químicos conocieran la secuencia de aminoácidos de una proteína, podrían predecir su estructura tridimensional. Esta idea era emocionante. Si lo conseguían, ya no tendrían que utilizar la cristalografía de rayos X y podrían ahorrar mucho tiempo. También podrían generar estructuras para todas las proteínas en las que no fuera aplicable la cristalografía de rayos X.
Estas conclusiones lógicas arrojaron el guante a lo que se ha convertido en el gran desafío de la bioquímica: el problema de la predicción. Para alentar un desarrollo más rápido en el campo, en 1994 los investigadores iniciaron un proyecto llamado Evaluación crítica de la predicción de la estructura de las proteínas (CASP, Critical Assessment of Techniques for Protein Structure Prediction), que se convirtió en una competición. Cada dos años, investigadores de todo el mundo tenían acceso a secuencias de aminoácidos en proteínas cuyas estructuras acababan de determinarse. Sin embargo, las estructuras se mantenían en secreto para los participantes. El desafío era predecir las estructuras de las proteínas basándose en las secuencias de aminoácidos conocidas.
CASP atrajo a muchos investigadores, pero resolver el problema de predicción resultó increíblemente difícil. La correspondencia entre las predicciones de los investigadores presentadas en el concurso y las estructuras reales apenas mejoró. El avance no se produjo hasta 2018, cuando un maestro de ajedrez, experto en neurociencia y pionero en inteligencia artificial entró en el campo.
El maestro de los juegos de mesa entra en las Olimpiadas de las Proteínas
Echemos un vistazo rápido a los antecedentes de Demis Hassabis: comenzó a jugar al ajedrez a los cuatro años y alcanzó el nivel de maestro a los 13 años. En su adolescencia, comenzó una carrera como programador y exitoso desarrollador de juegos. Comenzó a explorar la inteligencia artificial y se dedicó a la neurociencia, donde hizo varios descubrimientos revolucionarios. Usó lo que aprendió sobre el cerebro para desarrollar mejores redes neuronales para la IA. En 2010, cofundó DeepMind, una empresa que desarrolló modelos magistrales de IA para juegos de mesa populares. La empresa se vendió a Google en 2014 y, dos años después, DeepMind llamó la atención mundial cuando la empresa logró lo que muchos entonces creían que era el santo grial de la IA: vencer al campeón de uno de los juegos de mesa más antiguos del mundo, el Go.
Sin embargo, para Hassabis, el Go no era el objetivo, era el medio para desarrollar mejores modelos de IA. Tras esta victoria, su equipo estaba preparado para afrontar problemas de mayor importancia para la humanidad, por lo que en 2018 se inscribió en la decimotercera competición CASP.
Un triunfo inesperado para el modelo de IA de Demis Hassabis
En años anteriores, las estructuras proteínicas que los investigadores habían predicho para CASP habían alcanzado una precisión del 40 por ciento, en el mejor de los casos. Con su modelo de IA, AlphaFold, el equipo de Hassabis alcanzó casi el 60 por ciento. Ganaron y el excelente resultado sorprendió a mucha gente: fue un avance inesperado, pero la solución aún no era lo suficientemente buena. Para tener éxito, la predicción debía tener una precisión del 90 por ciento en comparación con la estructura objetivo.
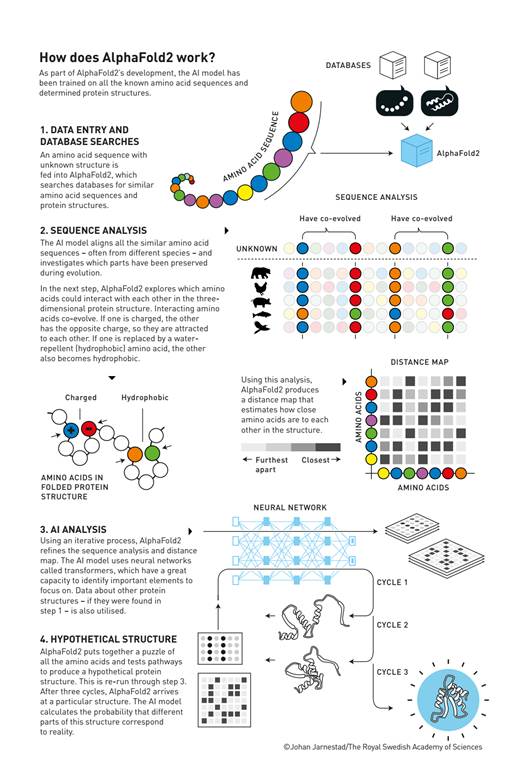
Figura 4. ¿Cómo funciona AlphaFold2?
La Figura 4 nos explica cómo trabaja AlphaFold2: Como parte del desarrollo de AlphaFold2, el modelo de IA ha sido entrenado en todas las secuencias de aminoácidos conocidas y las estructuras de proteínas determinadas.
1. Entrada de datos y búsquedas en bases de datos
Una secuencia de aminoácidos con estructura desconocida se introduce en AlphaFold2, que busca en bases de datos secuencias de aminoácidos y estructuras de proteínas similares.
2. Análisis de secuencias
El modelo de IA alinea todas las secuencias de aminoácidos similares, a menudo de especies diferentes, e investiga qué partes se han conservado durante la evolución. Con este análisis, AlphaFold2 produce un mapa de distancias que estima qué tan cerca están los aminoácidos entre sí en la estructura.
En el siguiente paso, AlphaFold2 explora qué aminoácidos podrían interactuar entre sí en la estructura tridimensional de la proteína. Los aminoácidos que interactúan coevolucionan. Si uno está cargado, el otro tiene la carga opuesta, por lo que se atraen entre sí. Si uno es reemplazado por un aminoácido repelente al agua (hidrofóbico), el otro también se vuelve hidrofóbico.
3. Análisis de IA
Con un proceso iterativo, AlphaFold2 refina el análisis de secuencias y el mapa de distancias. El modelo de IA utiliza redes neuronales llamadas transformadores, que tienen una gran capacidad para identificar elementos importantes en los que centrarse. También se utilizan datos sobre otras estructuras de proteínas, si se encontraron en el paso 1.
4. Estructura hipotética
AlphaFold2 arma un rompecabezas de todos los aminoácidos y prueba vías para producir una estructura proteica hipotética. Esto se vuelve a ejecutar hasta el paso 3. Después de tres ciclos, AlphaFold2 llega a una estructura particular. El modelo de IA calcula la probabilidad de que diferentes partes de esta estructura correspondan a la realidad.
Hassabis y su equipo continuaron desarrollando AlphaFold, pero, por mucho que lo intentaron, el algoritmo nunca llegó a funcionar. La dura verdad era que habían llegado a un punto muerto. El equipo estaba cansado, pero un empleado relativamente nuevo tenía ideas decisivas sobre cómo se podía mejorar el modelo de IA: John Jumper.
John Jumper acepta el gran reto de la bioquímica
La fascinación de John Jumper por el universo fue lo que le hizo empezar a estudiar física y matemáticas. Sin embargo, en 2008, cuando empezó a trabajar en una empresa que utilizaba superordenadores para simular proteínas y su dinámica, se dio cuenta que el conocimiento de la física podía ayudar a resolver problemas médicos.
Jumper llevó consigo este recién adquirido interés por las proteínas cuando, en 2011, comenzó su doctorado en física teórica. Para ahorrar capacidad informática, algo que escaseaba en la universidad, empezó a desarrollar métodos más sencillos e ingeniosos para simular la dinámica de las proteínas.
Pronto, él también aceptó el gran reto de la bioquímica. En 2017, había terminado recientemente su doctorado cuando oyó rumores de que Google DeepMind había empezado, en gran secreto, a predecir las estructuras de las proteínas. Les envió una solicitud de empleo. Su experiencia en simulación de proteínas le permitió tener ideas creativas sobre cómo mejorar AlphaFold, por lo que, después de que el equipo comenzó a mantenerse a flote, fue ascendido. Jumper y Hassabis codirigieron el trabajo que reformó fundamentalmente el modelo de IA.
Resultados sorprendentes con un modelo de IA reformado
La nueva versión, AlphaFold2, se inspiró en el conocimiento de Jumper sobre las proteínas. El equipo también comenzó a utilizar la innovación detrás del reciente y enorme avance en IA: las redes neuronales llamadas transformadores. Estas pueden encontrar patrones en enormes cantidades de datos de una manera más flexible que antes y determinar de manera eficiente en qué se debe enfocar para lograr un objetivo en particular.
El equipo entrenó a AlphaFold2 con la vasta información en las bases de datos de todas las estructuras de proteínas y secuencias de aminoácidos conocidas (Figura 4) y la nueva arquitectura de IA comenzó a ofrecer buenos resultados a tiempo para la decimocuarta competencia CASP.
En 2020, cuando los organizadores de CASP evaluaron los resultados, comprendieron que el desafío de 50 años de la bioquímica había terminado. En la mayoría de los casos, AlphaFold2 funcionó casi tan bien como la cristalografía de rayos X, lo cual fue asombroso. Cuando uno de los fundadores de CASP, John Moult, dio por concluida la competición el 4 de diciembre de 2020, se preguntó: ¿y ahora qué?

Figura 5. John Moult
Volveremos a ello. Ahora vamos a retroceder en el tiempo y arrojar luz sobre otro participante de CASP. Vamos a presentar la otra mitad del Premio Nobel de Química 2024, que trata sobre el arte de crear nuevas proteínas desde cero.
Un libro de texto sobre la célula hace que David Baker cambie de rumbo
Cuando David Baker empezó a estudiar en la Universidad de Harvard, eligió filosofía y ciencias sociales. Sin embargo, durante un curso de biología evolutiva se encontró con la primera edición del ya clásico libro de texto Molecular Biology of the Cell. Esto le llevó a cambiar de rumbo en la vida. Empezó a explorar la biología celular y, con el tiempo, se fascinó por las estructuras de las proteínas. Cuando, en 1993, empezó a ser jefe de grupo en la Universidad de Washington en Seattle, se enfrentó al gran reto de la bioquímica. Mediante ingeniosos experimentos, comenzó a explorar cómo se pliegan las proteínas. Esto le proporcionó conocimientos que llevó consigo cuando, a finales de los años 90, empezó a desarrollar un software informático que pudiera predecir las estructuras de las proteínas: Rosetta. Baker hizo su debut en la competencia CASP en 1998 usando Rosetta y, en comparación con otros participantes, le fue muy bien. Este éxito llevó a una nueva idea: que el equipo de David Baker podría usar el software a la inversa. En lugar de ingresar secuencias de aminoácidos en Rosetta y obtener estructuras de proteínas, deberían poder ingresar una estructura de proteína deseada y obtener sugerencias para su secuencia de aminoácidos, lo que les permitiría crear proteínas completamente nuevas.
Baker se convierte en constructor de proteínas
El campo del diseño de proteínas, en el que los investigadores crean proteínas a medida con nuevas funciones, comenzó a despegar a finales de los años 90. En muchos casos, los investigadores modificaron las proteínas existentes para que pudieran hacer cosas como descomponer sustancias peligrosas o funcionar como herramientas en la industria química. Sin embargo, la variedad de proteínas naturales es limitada. Para aumentar el potencial de obtener proteínas con funciones completamente nuevas, el grupo de investigación de Baker quería crearlas desde cero. Como dijo Baker, “Si quieres construir un avión, no empiezas modificando un pájaro; en lugar de eso, entiendes los primeros principios de la aerodinámica y construyes máquinas voladoras a partir de esos principios”.
Una proteína única ve la luz
El campo en el que se construyen proteínas completamente nuevas se llama diseño de novo. El grupo de investigación dibujó una proteína con una estructura completamente nueva y luego hizo que Rosetta calculara qué tipo de secuencia de aminoácidos podría dar como resultado la proteína deseada. Para ello, Rosetta buscó en una base de datos todas las estructuras proteínicas conocidas y buscó fragmentos cortos de proteínas que tuvieran similitudes con la estructura deseada. Utilizando el conocimiento fundamental del panorama energético de las proteínas, Rosetta optimizó estos fragmentos y propuso una secuencia de aminoácidos.
Para investigar el éxito del software, el grupo de investigación de Baker introdujo el gen para la secuencia de aminoácidos propuesta en bacterias que producían la proteína deseada. Luego determinaron la estructura de la proteína utilizando cristalografía de rayos X.
Resultó que Rosetta realmente podía construir proteínas. La proteína que los investigadores desarrollaron, Top7, tenía casi exactamente la estructura que habían diseñado (Figura 6).
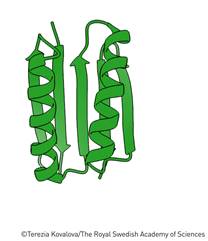
Figura 6. Top7: la primera proteína que era completamente diferente a todas las proteínas conocidas existentes
Espectaculares creaciones del laboratorio de Baker
Top7 fue una sorpresa para los investigadores que trabajaban en el diseño de proteínas. Los que habían creado proteínas de novo anteriormente solo habían podido imitar estructuras existentes. La estructura única de Top7 no existía en la naturaleza. Además, con sus 93 aminoácidos, la proteína era más grande que cualquier otra producida anteriormente mediante diseño de novo.
Baker publicó su descubrimiento en 2003. Este fue el primer paso en algo que solo puede describirse como un desarrollo extraordinario; algunas de las muchas proteínas espectaculares creadas en el laboratorio de Baker se pueden ver en la Figura 7. También publicó el código para Rosetta, por lo que una comunidad de investigación global ha seguido desarrollando el software, encontrando nuevas áreas de aplicación.
Es hora de atar los cabos sueltos del Premio Nobel de Química 2024. ¿Y ahora qué?
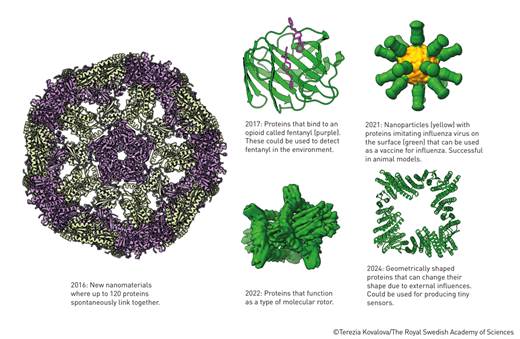
Figura 7. Proteínas desarrolladas utilizando el programa Rosetta de Baker
En la figura 7 se incluyen las proteínas creadas en el laboratorio de Baker:
2016: Nuevos nanomateriales en los que hasta 120 proteínas se unen espontáneamente.
2017: Proteínas que se unen a un opioide llamado fentanilo (violeta). Podrían utilizarse para detectar el fentanilo en el medio ambiente.
2021: Nanopartículas (amarillas) con proteínas que imitan al virus de la influenza en la superficie (verde) que pueden utilizarse como vacuna contra la influenza. Se han obtenido resultados exitosos en modelos animales.
2022: Proteínas que funcionan como un tipo de rotor molecular.
2024: Proteínas con forma geométrica que pueden cambiar su forma debido a influencias externas. Podrían utilizarse para producir sensores diminutos.
El trabajo que antes llevaba años ahora se realiza en tan solo unos minutos
Cuando Demis Hassabis y John Jumper confirmaron que AlphaFold2 realmente funcionaba, calcularon la estructura de todas las proteínas humanas. Luego predijeron la estructura de prácticamente todos los 200 millones de proteínas que los investigadores han descubierto hasta ahora al mapear los organismos de la Tierra.
Google DeepMind también ha puesto a disposición del público el código de AlphaFold2 y cualquiera puede acceder a él. El modelo de IA se ha convertido en una mina de oro para los investigadores. Para octubre de 2024, AlphaFold2 había sido utilizado por más de dos millones de personas de 190 países. Antes, a menudo se necesitaban años para obtener la estructura de una proteína, si es que se conseguía. Ahora se puede hacer en unos minutos. El modelo de IA no es perfecto, pero estima la exactitud de la estructura que ha producido, por lo que los investigadores saben cuán confiable es la predicción. La Figura 5 muestra algunos de los muchos ejemplos de cómo AlphaFold2 ayuda a los investigadores.
Después de la competencia CASP 2020, cuando David Baker se dio cuenta del potencial de los modelos de IA basados en transformadores, agregó uno a Rosetta, lo que también facilitó el diseño de novo de proteínas. En los últimos años, del laboratorio de Baker han surgido una increíble creación de proteínas una tras otra (Figura 7).
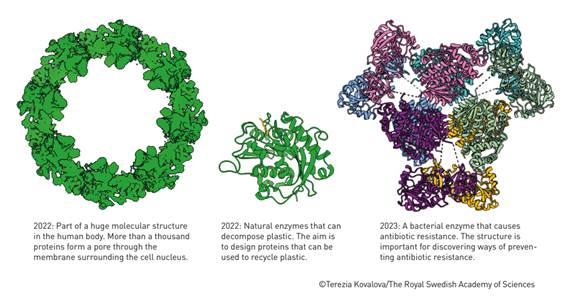
Figura 8. Ejemplos de estructuras de proteínas determinadas utilizando AlphaFold
2022: Parte de una enorme estructura molecular en el cuerpo humano. Más de mil proteínas forman un poro a través de la membrana que rodea el núcleo celular.
2022: Enzimas naturales que pueden descomponer el plástico. El objetivo es diseñar proteínas que puedan usarse para reciclar el plástico.
2023: Una enzima bacteriana que causa resistencia a los antibióticos. La estructura es importante para descubrir formas de prevenir la resistencia a los antibióticos
Desarrollo vertiginoso en beneficio de la humanidad
La asombrosa versatilidad de las proteínas como herramientas químicas se refleja en la enorme diversidad de la vida. El hecho de que ahora podamos visualizar tan fácilmente la estructura de estas pequeñas máquinas moleculares es alucinante; nos permite comprender mejor cómo funciona la vida, incluyendo por qué se desarrollan algunas enfermedades, cómo se produce la resistencia a los antibióticos o por qué algunos microbios pueden descomponer el plástico.
La capacidad de crear proteínas cargadas de nuevas funciones es igualmente asombrosa. Esto puede conducir a nuevos nanomateriales, fármacos específicos, un desarrollo más rápido de vacunas, sensores mínimos y una industria química más ecológica, por nombrar solo algunas aplicaciones que son para el mayor beneficio de la humanidad.